
Genomics and Bioinformatics Active
Genetic analysis is increasingly used to understand ecosystem processes and inform conservation, management, and policy. I assist USGS researchers and their collaborators in the design, analysis, and interpretation of high-throughput genetic studies. Common applications include: detecting genes responsive to particular environmental stressors in a sentinel species or species of conservation concern; generating reference genome sequences of pathogens for functional or evolutionary analysis; identifying genetic variation that distinguish populations or species; using “barcode” sequences to identify species in gut contents, feces, or environmental samples.
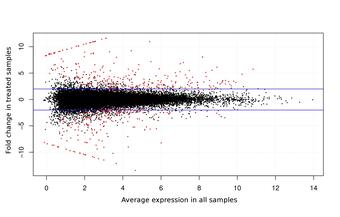
Transcriptomics
Various RNAs are transcribed from the genome, including the mRNAs that encode proteins. Isolating the mRNA from a tissue sample and sequencing it with high-throughput technology allows the underlying “coding sequence” of the genome to be reconstructed and compared to databases of known sequences to infer potential functions. It also allows the statistical analysis of differential gene expression between two or more sample types, such as experimentally manipulated cohorts and the corresponding control cohort. Such an experiment may establish the physiological relevance of a potential stressor, or identify candidate biomarkers that provide early warning of those stressors before irreparable harm is done to a sensitive population.
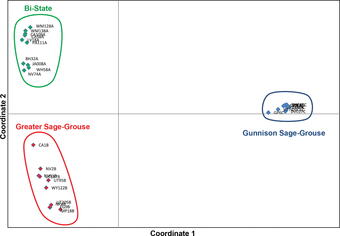
Population Genomics
The genomes of most organisms are very similar within a given species, with increasing levels of divergence accruing at higher taxonomic ranks reflecting longer divergence times. However, most genomes contain millions to billions of bases of DNA, so that even if only 1 in 100,000 bases varies within a species, individuals may still differ from conspecifics at thousands of genomic positions. High-throughput sequencing facilitates the identification of these variable sites and estimation of the frequencies of the different variants. This information can be used to construct well-supported species phylogenies, estimate gene flow or hybridization events, and detect non-neutral patterns of evolution. This type of information is critical for effective implementation of conservation measures.
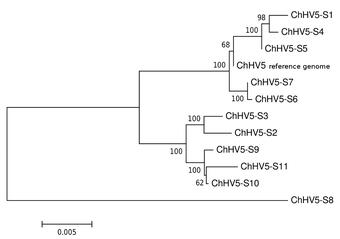
Metagenomics
Metagenomics is the analysis of genomic fragments in complex mixtures that are inherently difficult to separate by species, such as microbial communities or a host tissue infected with a virus. High-throughput sequencing and computational approaches are used to identify the microbial taxa present, the genes they harbor (such as toxins or antibiotics), and the complex interaction between pathogen and host that might lead to disease, for example.
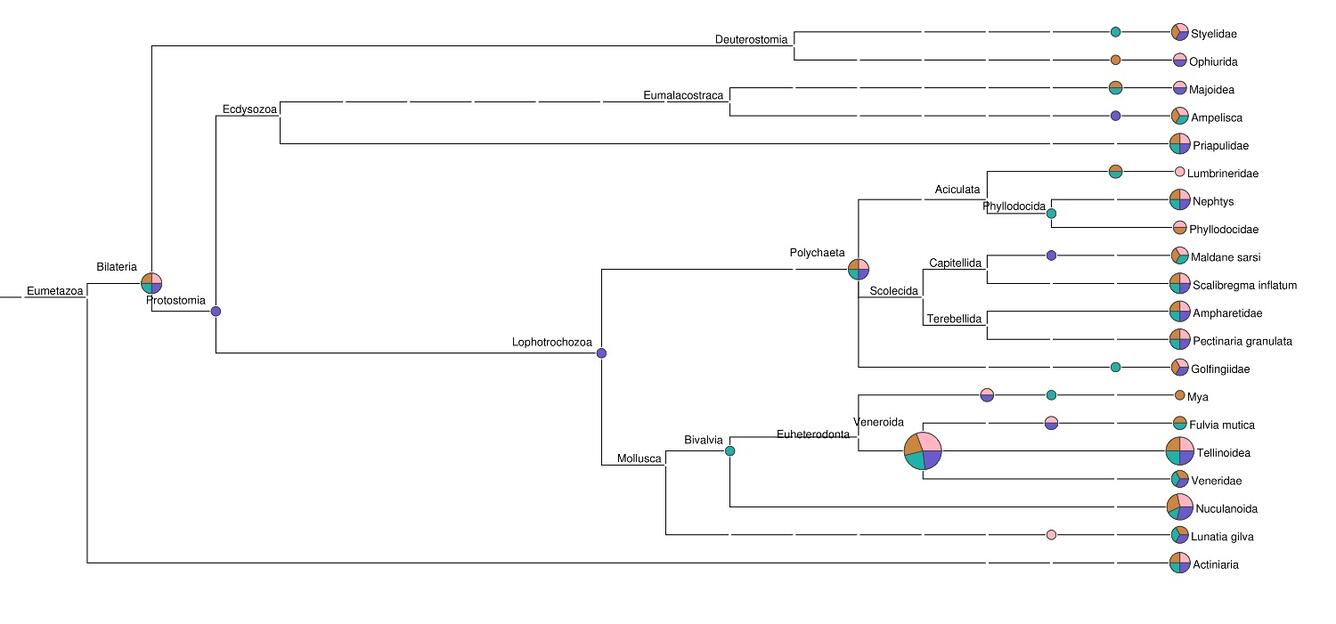
Barcode Sequencing
Genetic “barcodes” are unique signatures that reveal the species from which detected DNA derives. Barcode sequencing complements metagenomics, differing in that only certain small regions that are most informative of biological origin are examined, rather than the entire “metagenome” of the sample. Barcode methods can be used to reconstruct the diet of animal species noninvasively from feces, quantify the biodiversity of minute larvae dispersing in water, or detect invasive plant species with a pollen trap.
Genetic analysis is increasingly used to understand ecosystem processes and inform conservation, management, and policy. I assist USGS researchers and their collaborators in the design, analysis, and interpretation of high-throughput genetic studies. Common applications include: detecting genes responsive to particular environmental stressors in a sentinel species or species of conservation concern; generating reference genome sequences of pathogens for functional or evolutionary analysis; identifying genetic variation that distinguish populations or species; using “barcode” sequences to identify species in gut contents, feces, or environmental samples.
Transcriptomics
Various RNAs are transcribed from the genome, including the mRNAs that encode proteins. Isolating the mRNA from a tissue sample and sequencing it with high-throughput technology allows the underlying “coding sequence” of the genome to be reconstructed and compared to databases of known sequences to infer potential functions. It also allows the statistical analysis of differential gene expression between two or more sample types, such as experimentally manipulated cohorts and the corresponding control cohort. Such an experiment may establish the physiological relevance of a potential stressor, or identify candidate biomarkers that provide early warning of those stressors before irreparable harm is done to a sensitive population.
Population Genomics
The genomes of most organisms are very similar within a given species, with increasing levels of divergence accruing at higher taxonomic ranks reflecting longer divergence times. However, most genomes contain millions to billions of bases of DNA, so that even if only 1 in 100,000 bases varies within a species, individuals may still differ from conspecifics at thousands of genomic positions. High-throughput sequencing facilitates the identification of these variable sites and estimation of the frequencies of the different variants. This information can be used to construct well-supported species phylogenies, estimate gene flow or hybridization events, and detect non-neutral patterns of evolution. This type of information is critical for effective implementation of conservation measures.
Metagenomics
Metagenomics is the analysis of genomic fragments in complex mixtures that are inherently difficult to separate by species, such as microbial communities or a host tissue infected with a virus. High-throughput sequencing and computational approaches are used to identify the microbial taxa present, the genes they harbor (such as toxins or antibiotics), and the complex interaction between pathogen and host that might lead to disease, for example.
Barcode Sequencing
Genetic “barcodes” are unique signatures that reveal the species from which detected DNA derives. Barcode sequencing complements metagenomics, differing in that only certain small regions that are most informative of biological origin are examined, rather than the entire “metagenome” of the sample. Barcode methods can be used to reconstruct the diet of animal species noninvasively from feces, quantify the biodiversity of minute larvae dispersing in water, or detect invasive plant species with a pollen trap.